

Grounding Perturbation Prediction in Biological Knowledge
Decoding Transcriptomics Responses with TxPert

tl;dr: TxPert is a lightweight deep learning framework that predicts how cells respond transcriptomically to genetic perturbations, including gene combinations never seen during training. It integrates curated biological knowledge graphs as structural guidance, forcing the model to learn actual biology instead of statistical artifacts. TxPert approaches split-half experimental reproducibility on single perturbations, outperforms existing models on double perturbations, and generalizes to entirely unseen cell lines. We also surface a methodological contribution: widely used benchmarks have been inflating perceived model performance across the field, and we introduce stricter evaluation standards in addition to more descriptive baselines and ceilings. Mastering transcriptomic prediction is a prerequisite for the larger goal of simulating full cellular behavior across modalities, and TxPert is one step toward making that a reality.
Connect with us: Valence is constantly seeking talented individuals with diverse backgrounds and expertise to join our team. Explore open roles here.
Scaling Phenotypic Discovery Beyond the Lab
Historically, the computational toolkit for drug discovery has favored target-based approaches: identifying a specific protein hypothesized to be involved in a disease and designing a molecule to bind to it. Phenotypic drug discovery, by contrast, looks at the holistic, observable changes in a cell. It asks a broader question: Regardless of the specific target, does this intervention make a sick cell look and act like a healthy one? Today, scientists have access to incredibly powerful tools to digitize and scale this phenotypic discovery process. By using CRISPR technology to introduce genetic perturbations, researchers can use gene editing as a proxy of diseased state for chemical interventions. CRISPR acts as molecular scissors, allowing scientists to systematically knock down or overexpress specific genes and observe how the cell responds. Researchers then capture the cell’s holistic response in the form of machine-readable, high-dimensional data. This includes approaches like microscopy imaging readouts that show the physical characteristics of the cell and or single-cell transcriptomics. Transcriptomics is particularly powerful because it reads the messenger RNA within the cell, revealing exactly which genes are turned on or off at any given moment, providing a comprehensive functional snapshot of the cellular state.
Systematically mapping these observable changes is at the core of our approach to drug discovery at Recursion. However, exhaustively testing all possible multi-gene perturbations across human tissues in a wet lab is prohibitively expensive and time-consuming. Consider the sheer combinatorial scale of the problem: humans have roughly 20,000 protein coding genes, and exploring all possible pairs of genes to understand double perturbations requires nearly 200 million unique combinations. Multiply that by hundreds of distinct cell types in the human body, and you quickly reach tens of billions of required experiments. This is a scale that physical laboratories, no matter how automated, simply cannot accommodate. To overcome this, we must translate physical biology into computational simulations. We need to explore and understand complex cellular behaviors in silico to predict outcomes at scale and discover novel, actionable biology.
The Data Bottleneck: Why Current SOTA Models Fail
As the AI community pushes toward cellular simulation, independent benchmarking studies have made one thing clear: many high-profile transcriptomics foundation models underperform what was expected of them.
The core issue stems from the illusion of infinite data. While biological data is increasingly available, the lack of diversified conditions contained within both public and proprietary datasets means that available data is barely scratching the surface of capturing true biological complexity. Imagine taking a million photographs of the exact same street corner; you have a massive amount of data, but you have learned very little about the rest of the world. Using transcriptomic data alone, training these massive models with the aim of generalizing across all of biology, in an unsupervised manner, is currently a long shot to achieve given the current lack of biological diversity.
Many current state-of-the-art models fail because they lack the structural guidance needed to navigate this incredibly sparse and noisy data landscape. In biology, prior knowledge is a necessary modality. Humanity has spent decades meticulously building curated databases detailing which genes interact with one another and how biochemical pathways function. This accumulated human knowledge is not an optional crutch for an algorithm; it is an absolute necessity to ground computational models in causal biological rules.
By explicitly feeding a model proven biological rules, we drastically reduce its reliance on data scaling. More importantly, we prevent the deep learning model from overfitting to simple, non-causal correlations found within limited training sets instead forcing the model to learn actual biology rather than statistical artifacts. While this choice might introduce new limitations given how current biological rules are limited by our current understanding, it temporarily bridges the data diversity gap until more data is available.
TxPert: A Principled Method for Integrating Prior Knowledge
To demonstrate this, we developed TxPert: a latent deep learning framework designed explicitly to predict transcriptomic responses to unseen genetic perturbations by bridging simple deep learning with established biology.
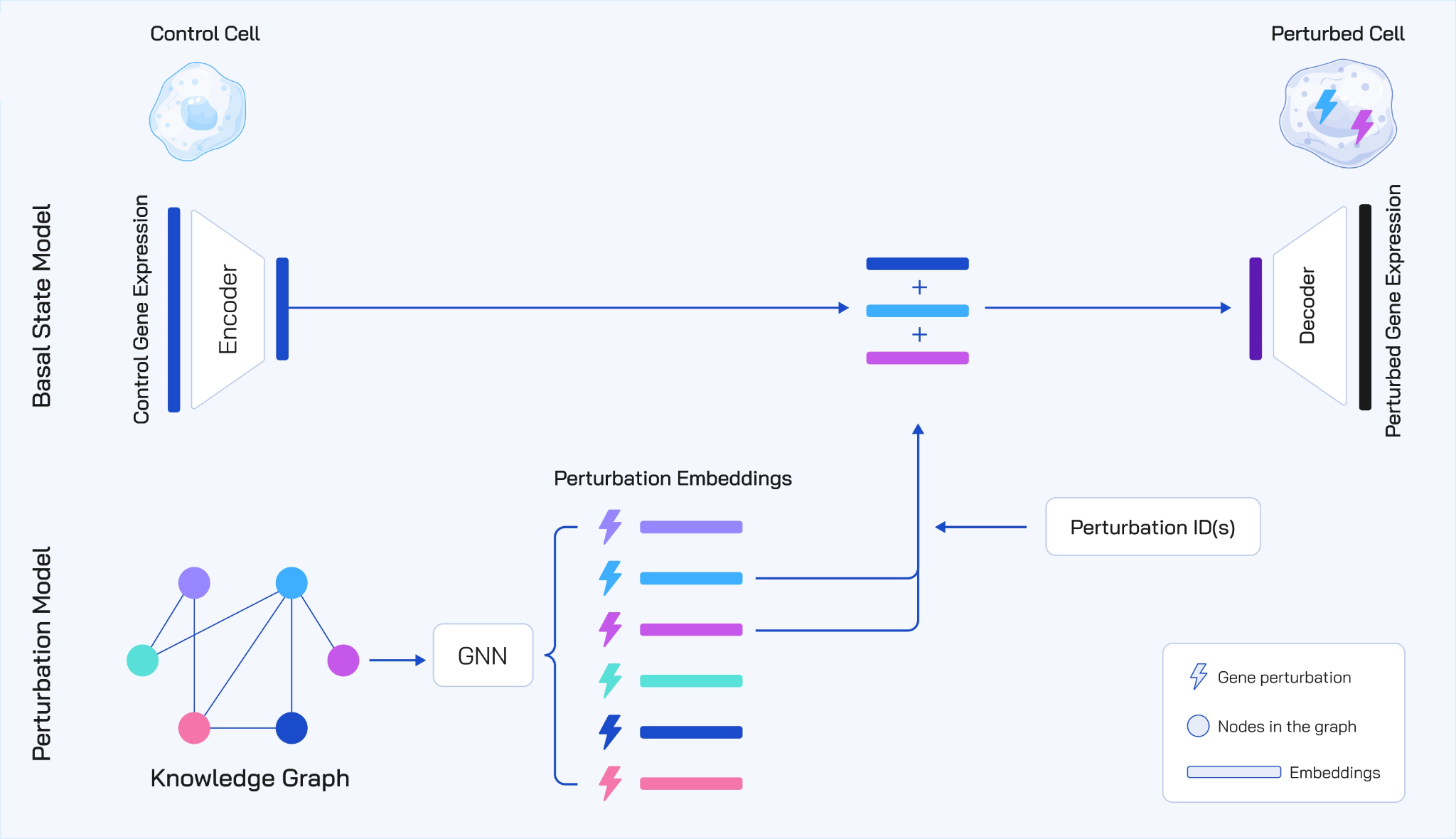
Figure 1: To avoid data bottlenecks and overfitting, TxPert utilizes a lightweight, inherently efficient Multilayer Perceptron (MLP) to encode the basal state from control expression. Contributions are focused on the Perturbation Encoder, which employs Graph Neural Networks (GNNs) utilizing bidirectional message passing and expander graph attention to ingest diverse knowledge graphs from public and internal Recursion sources (STRING, PxMap, TxMap). A transition model integrates these embeddings to decode the final predicted perturbed gene expression. The total parameter size is 8 million.
“TxPert outperforms through deliberate simplicity”
Instead of using massive compute-heavy transformer models to encode the unperturbed control cell, which we call the basal state, TxPert relies on an inherently efficient, simple Multilayer Perceptron. This Multilayer Perceptron is a foundational neural network architecture that is computationally efficient while still retaining the capacity to capture the complex, nonlinear dependencies inherent in gene expression data. The model saves its computational complexity for where it matters most: mapping the complex effects of the perturbations. It maps the high-dimensional gene expression of a control cell into a compact, low-dimensional latent embedding space.
We found that the specifics of how we integrate prior knowledge matter a lot. We tested multiple Graph Neural Network architectures to encode the genetic perturbations. Graph Neural Networks are uniquely suited for this task because they treat biological entities like genes as nodes and their biochemical interactions as the connecting edges. The most successful approaches we discovered were surprisingly straightforward:
Hybrid Bi-Directional Message Passing: This is a simple message passing model that explicitly aggregates information from both incoming and outgoing edges of a perturbation. In biology, a gene does not act in isolation. It is influenced by upstream regulators and, in turn, influences downstream targets. By looking in both directions, this architecture allows the model to capture the full biological ripple effect, learning from what influences the perturbed gene and vice versa.
Exphormer MG: This is a multi graph transformer that efficiently handles complex, long range dependencies across incomplete biological graphs. Traditional graph models often struggle when nodes are far apart. The Exphormer architecture builds a sparse attention pattern by combining the original biological graph edges with edges from an expander graph. An expander graph acts like a system of wormholes, providing shortcut paths that improve information flow and allow the network to fill in the gaps where our biological knowledge might be missing.
Different scientific databases represent different views of biology. To maximize predictive power, TxPert combines curated literature graphs like STRING and Gene Ontology with graphs derived from our proprietary maps of biology developed at Recursion, specifically the PxMap and TxMap that are based on high-throughput Phenomics and Transcriptomics screens, respectively.
To definitively prove that this biological prior knowledge was actually contributing to performance, we performed rigorous ablations. When we randomly rewired the edges of the STRING graph, or progressively deleted them, the predictive accuracy of the model directly and consistently collapsed. TxPert was not just memorizing the training data; rather, it was heavily relying on the structural biological rules we had provided to make its predictions.
The Benchmarking Reality Check: Fixing Flawed Evaluations
Before we could further evaluate the capabilities of TxPert, we had to address one of the longstanding challenges in computational biology: even when deep learning models seem to perform well, the benchmarks themselves are often fundamentally flawed.
This was evident in prior work in which we demonstrated that a simple Principal Component Analysis on transcriptomics data could outperform complex foundation models on perturbation representation learning benchmarks. Principal Component Analysis is a classical statistical technique used to reduce dimensionality, which suggests a need to revisit evaluation metrics following outperformance of modern AI methods.
Additional prior work has shown that a non-learned mean baseline often predicts individual perturbation responses better than prominent SOTA approaches. The mean baseline is a simple approach averaging the transcriptomic effects of all perturbations in the training set, and guessing that exact same average for any new, unseen perturbation.
How could simple mathematical averages beat SOTA approaches? It’s because the mean baseline appears to represent a defining characteristic of biological data. When a cell is genetically perturbed, especially when an essential gene is targeted, it undergoes general cellular stress. The cell shifts its resources from active growth to a state of quiescence and recycling. For example, the cell upregulates lysosomes, which act as the waste disposal system of the cell, and strongly downregulates the complex machinery required for cell division. Complex deep learning models were simply getting credit for memorizing and regurgitating this generic, universal stress response rather than learning the specific, unique functional effect of the intended perturbation itself, as evaluated in depth in the Systema study.
Additionally, evaluating models without sufficient batch matched controls can result in mistaking background experimental noise for true perturbation effects. Experimental batch effects are a well-known challenge in biological data where cellular states vary due to the extreme sensitivity of biological systems and their subtle interactions with the lab environment. Confounding cannot be strictly controlled because biological processes like transfection and survival are inherently stochastic. Failing to account for these batch effects artificially inflates perceived model performance by adding avoidable noise.
To combat this, we explicitly designed our metrics to isolate true biological signals. We focus on the Pearson delta metric, which calculates the correlation between the predicted and observed log fold change versus the batch-matched control mean. We complement this with retrieval metrics, which focus on relative similarity, capturing whether a prediction for any given perturbation is more similar to the true profile of this perturbation or to other perturbations; and thus if a model can distinguish perturbations or is just capturing systemic effects.
To contextualize what actually constitutes genuinely useful computational performance, we introduced a much harder, unforgiving baseline of split-half experimental reproducibility. We aim to ask, given the inherent and unavoidable noise in biology, whether physical replicates of the exact same biological experiment predict each other? We calculate this by dividing test cells into two roughly equal halves, calculating the mean expression profiles for each half, and measuring the agreement between these means. This provides a rigorous, human-level reference point, representing not the absolute best a model could achieve, but a competitive mark to beat. This would be a somewhat similar proxy to the performance achieved by paying for and executing half the experiments.
Promising Results & Drug Discovery Impact
When evaluated against these newly defined baselines across several out-of-distribution tasks, TxPert delivered exciting results.
Approaching the Experimental Ceiling (Single Unseen Perturbations): When predicting the complex transcriptomic effects of single genetic perturbations not seen during training, TxPert achieves SOTA performance. It approaches split half experimental reproducibility, meaning TxPert is effectively predicting cellular outcomes as well as conducting real-world lab experiments.
Solving Combinatorial Costs (Double Perturbations): Testing multi-gene perturbations physically is where laboratory costs scale exponentially. TxPert successfully predicts the transcriptomic effects of novel double gene perturbations. In doing so, it outperforms existing literature models like GEARS and scLAMBDA, while also outperforming simple additive mathematical baselines.
Generalizing across contexts (Cross-Cell Line): In a test explicitly designed for real-world drug discovery applications, TxPert is able to predict perturbation outcomes in entirely unseen cell lines. This means absolutely no perturbations in these specific target cell lines were ever observed by the model during its training phase. TxPert beats most alternative baseline methods in this difficult, cross context task.
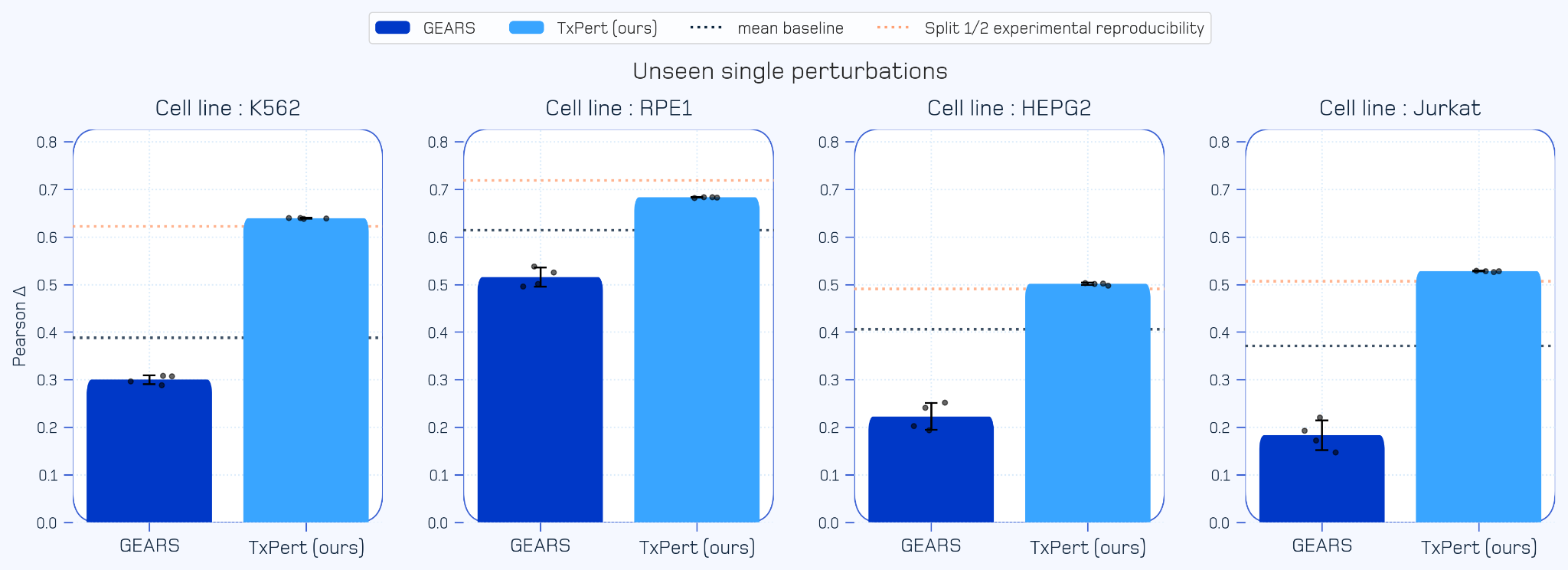
Figure 2: TxPert performance on predicting unseen single perturbations. This chart evaluates predictive accuracy using the Pearson Delta metric across four distinct cell lines. TxPert outperforms both SOTA approaches and simple mean baseline. TxPert also matches or approaches the strict upper limit of split half experimental reproducibility.
This work, alongside other recent approaches like CellFlow, STATE and X-Cell, hold immense promise for how drug discovery could be done. It unlocks the potential to perform highly targeted confirmatory screens, turning the lab from a search tool into a validation tool, directing limited laboratory resources to the areas of biology where they’re needed most and avoiding experiments where outcomes can already be confidently predicted.
Where This Fits, and What’s Next
The success of TxPert highlights a philosophical discussion currently being explored in the computational biology field. Models like STATE attempt to learn the complex mappings between control and perturbed cellular distributions from scratch, relying on large amounts of data. While data driven scaling has been proven to solve many bottlenecks in many domains, the current severe lack of contextual data diversity in biology makes the integration of biological prior knowledge a very useful addition to bridge the data gap. At the same time, recent data generation initiatives such as the Biohub $500M initiative hold a lot of potential for exploiting scaling laws and expanding biological data diversity.
Recent community benchmarks (such as the GenBio Benchmark) reaffirm what we establish with TxPert: true experimental accuracy represents a hard mathematical ceiling, and simple non-learned baselines are difficult to beat once you evaluate models correctly. TxPert provides the foundational blueprint for how to actually cross that difficult threshold using principled engineering.
This work also serves as a crucial reminder that computational scale is a tool, not an end goal. Scaling model parameters and computing power can be very valuable when it actively drives down the staggering costs of drug discovery, or unlocks genuinely new predictive capabilities.
Going Forward: Accurately predicting transcriptomic perturbations is one piece of a much larger problem. However, progress on modelling single omics (transcriptomics being a complex and high-dimensional data modality in its own right) is a prerequisite to unlocking additional downstream use cases and impact on increasingly complex biological systems across multiple modalities. We are encouraged to see that the field is already devoting significant resources towards combining transcriptomics with simulating microscopy imaging, understanding spatial tissue context, and simulating proteins, all with the aim of advancing our understanding of biology.
Thank you to Valence researchers Ali Denton and Frederik Wenkel for their thoughtful feedback and contributions to this piece.
This post is part of “Inside Valence”, a series where you’ll get a behind-the-scenes look at our research, exploring new ways to predict, explain, and ultimately decode biology. If this resonates, consider subscribing!
 | A guest post by
|