

Truly All-Atom Boltzmann sampling with AquaGen


Connect with us: Valence is hosting a social event on July 10th at ICML in Seoul, Korea. If you would like to meet our team and others working at the frontier of AI x drug discovery, you can register here.
In this blog post, we’re excited to present AquaGen, the first all-atom, explicit solvent, periodic-boundary-condition-aware generative model that produces molecular configurations from the Boltzmann distribution at a fraction of the cost of molecular dynamics (MD). This enables the prediction of molecular properties, such as absolute hydration free energy (AHFE), in a gray box manner: as ensemble averages of force field evaluations over the generated samples. We believe this is an important stepping stone in moving the field of drug discovery from ML predictions that are often unreliable and unexplainable in practice, to predictions that are inspectable, grounded in physics, refinable via additional MD simulations, have calibrated uncertainties, and benefit from test-time compute scaling. This post is intended to serve as a high-level, accessible introduction to AquaGen, and we encourage you to check out the full paper here.
Molecular Dynamics, Forces, and Energy
Imagine you wanted to better understand something about a molecule, say, what happens to sugar when you put it in your coffee, at a very basic level. Computational chemists figured out that the best way to do this is often to just model every atom in a system that you care about, and then, like pressing play on a movie, letting the equations of physics play the movie of all the atoms dancing in space.
This is usually done by asking a simple question: for a given 3D arrangement of atoms in space, how much force do they exert on each other? You may remember F=ma from high school, or Coulomb’s law, F=k q1q2/r2, and Hooke’s law for springs. What modern simulators use isn’t all that different in spirit, although decades of research have been devoted to making these force-fields as accurate and efficient as possible (of course, if you’ve ever learned about quantum mechanics or quantum field theory, you would know that thinking about atoms like little charged balls is a cartoonish representation of reality, but for many questions, force-fields give a sufficient answer).
Using these forces, which are 3D vectors, we’re able to tell in which direction each atom wants to go. This allows simulators to take tiny steps, and I really mean tiny: each step is typically one or a few femtoseconds (that’s 10^-15 seconds). If we take a lot of these steps, all while modeling temperature as extra noise on atom movement, we’re doing what’s called Molecular Dynamics (MD). Taking millions of steps to simulate a few nanoseconds to microseconds (10^-9 to 10^-6 seconds) can be enough for us to understand a lot about many molecular systems. The longer we run a simulation, the more different states of the system we end up seeing. If we were to run this for very very long, the distribution of states that we would recover is known as a Boltzmann distribution, which says that the probability of visiting a state becomes (exponentially) less likely as the energy of that state increases.
In some sense, energy is a more fundamental quantity than forces. You can think of energy as how unhappy a group of atoms is. Very high energy, and the atoms are desperate to find a different arrangement (imagine lying upside down on your couch, with your head on the floor and your feet up on the headrest; that’s a high energy position that your body would probably want to change). Low energy, and the system doesn’t want to change. It also happens to be that energy and forces are cleanly related: forces are the negative gradient of the energy with respect to positions.
Solvents and Free Energy
In drug discovery, an important ingredient is the solvent. Biology tends to happen not in a vacuum, but in water. Indeed, getting accurate predictions for hydration, solvation, permeability, binding, or pose diversity–all important ways to understand drug molecules–crucially depend on simulating molecules in water. From a modelling point of view, this is perhaps the most significant difference to prior work we introduce in AquaGen: explicitly modeling water.
Here, it’s important to mention the concept of free energy. Think of free energy as an expanded notion of energy that takes into account how much freedom exists in a given state. Typically, we care more about free energy differences between two states, rather than absolute free energy. If someone was actually holding you upside down on the couch, and then liberated you, your difference in happiness before and after (combined with how much more mobile you are when not upside down) would be the free energy difference associated with that change. When you pour that sugar in your coffee, sugar molecules spread around because they are happier when spread around and cohabitating with water molecules than when stuck with other sugar molecules in your sugar cube crystal. In other words, there is a negative (favorable) free energy difference associated with this process.
In the context of drug discovery and solvents, we often care about the free energy difference between a drug-like molecule solvated in water, and that same molecule in vacuum. This is known as the absolute hydration free energy (AHFE), and it is one of many important quantities that can be computed from MD simulations (while the words “absolute” and “free energy” appear in AHFE, it’s important to remember that it is still a difference in free energy between two states)
Generative modeling with machine learning
Another, often faster, way to generate distributions of states, necessary for computing quantities like free energies, is to use machine learning. If you’ve ever used generative AI, may it be chat bots, image, or video generators, you’ve used ML models trained on essentially the same principle: transforming random noise into samples from a particular data distribution.
It turns out that deep learning models can be surprisingly good at answering the following question: “given this very corrupted input (think, a random RGB image) what would the clean version look like?”. Using the answer to this question, which comes in the form of a velocity field, but only taking little steps one at a time, generative ML models are able to reproduce their training data to a high degree of fidelity.
This method has an interesting property: every time we want to generate a new state, we start from a new sample of total noise, and end up with a fresh new independent state. In terms of MD, it means that we no longer have to sequentially “play the movie” of physics for a long time to get many different physical poses of the system. Instead we just draw anew! This is a common approach in ML, and using models to draw diverse samples of molecular conformations (i.e. the Boltzmann distribution) enables a more global understanding of systems without iterative timestepping.
This is exactly what we do in AquaGen, at scale. Using data from MD, we train a model to transform noise into 3D arrangements of the thousands of atoms that make up drug-like molecules and water. The model itself is a standard 3D graph neural network which simply sees atom types & properties, as well as local k-NN distances and vectors between atoms. After we generate many of these 3D arrangements for a given drug-like molecule, we can compute the energy of each one, and use standard physics techniques to compute the desired free energy differences.
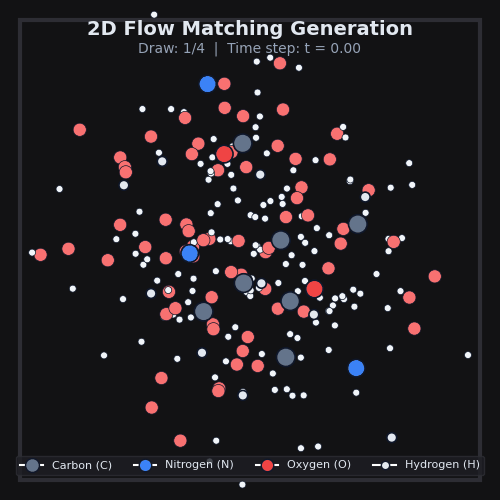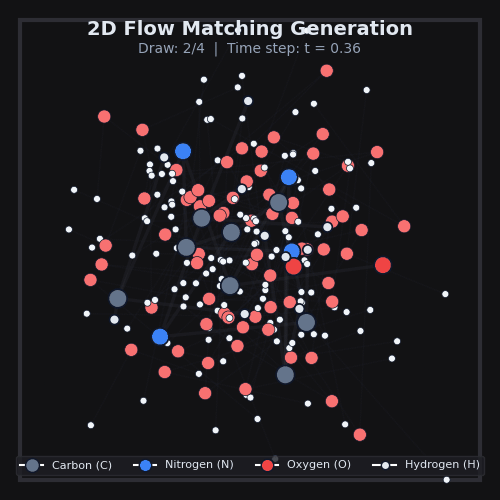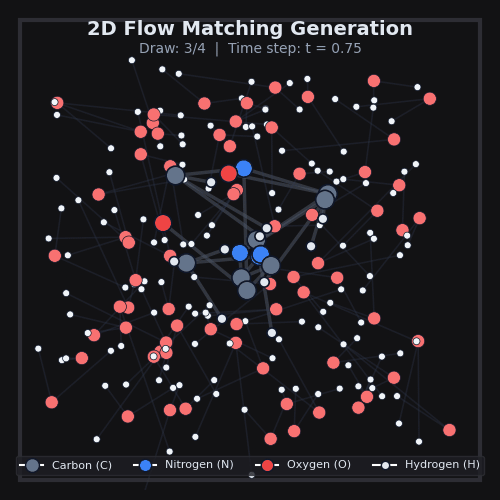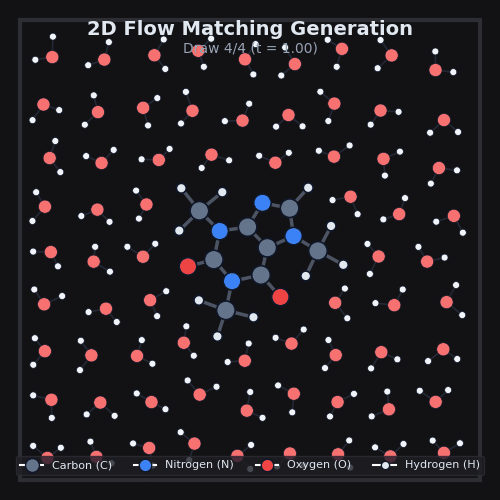
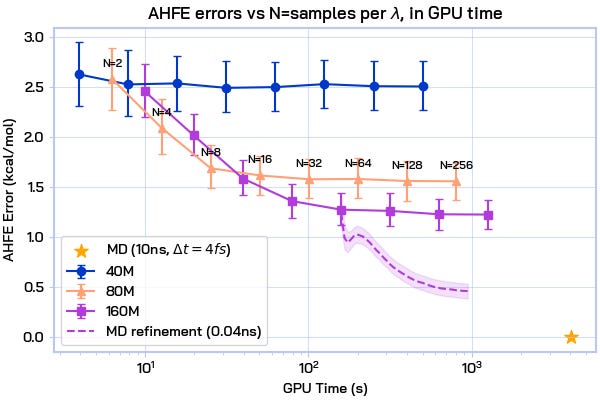
AquaGen is fast: because we benefit from this independent sampling and because we only need a few hundred samples of a molecule in water to get good estimates of the hydration free energy, we’re able to obtain accurate (~1kcal/mol error with respect to an MD reference) answers about 10x faster than MD (which has to, by design, do millions of steps).
AquaGen is refinable: because we generate the full picture, i.e. every H2O and compound atom, we’re trivially able to plug the outputs of AquaGen directly into an MD simulation, which in a few thousand steps fixes many small errors that our GNN model is making, without adding much overhead. This gets our free energy errors down below 0.5 kcal/mol.
AquaGen is interpretable & grounded: again because we generate full 3D systems, it is quite easy to simply look at what the model produces and reason “in physical space” about what may be going wrong. We used this property throughout the development of AquaGen, and we anticipate it being key to future improvements. More fundamentally, AquaGen is not a black box model: the GNN does not predict a free energy estimate, instead, we use force fields to evaluate the energy of the 3D arrangements produced by our GNN, and use those energies in a gray box manner to compute a free energy prediction. This grounds the predictions that we make in the physics that we emulate, and makes it much harder for the model to cheat and hallucinate answers.
Finally, AquaGen is calibrated: as a result of having gray box estimates, we have, almost for free, the very useful ability to compute the uncertainty of our prediction by bootstrapping subsamples to obtain confidence intervals. In practice, these uncertainties correlate very well with the actual errors that the model makes.
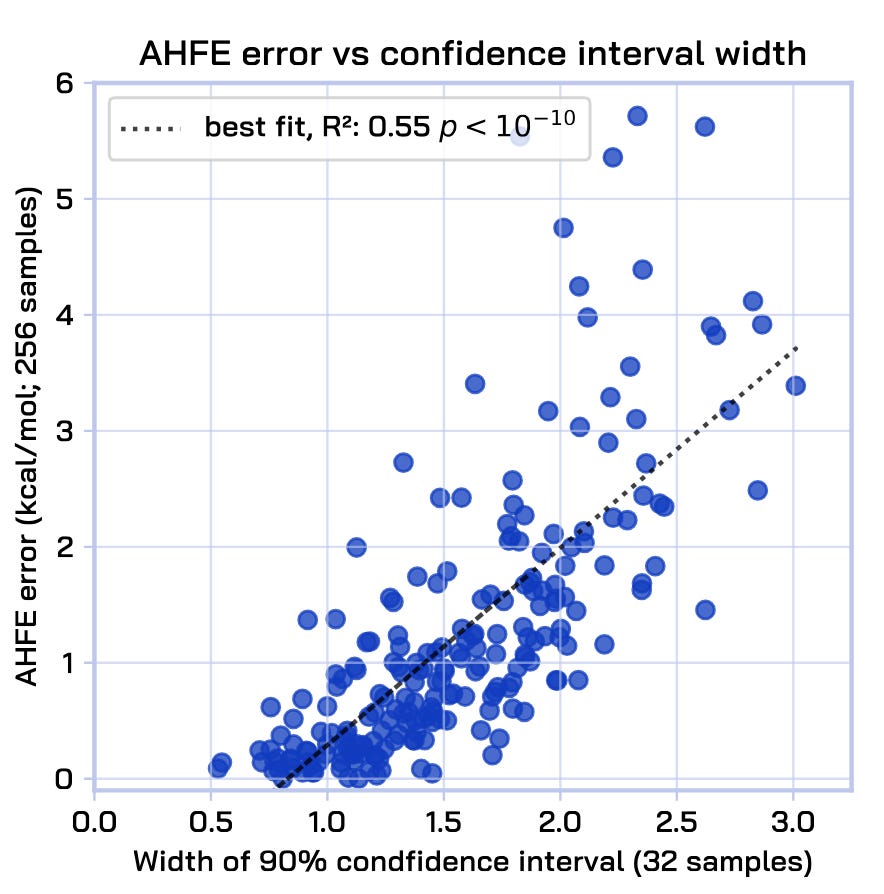
The combination of all of these ingredients–independent samples, physical groundedness, refinability, and calibration–enables arbitrary test-time compute scaling: more independent samples can be drawn as needed, and they can be refined as needed until a satisfactory accuracy and confidence is reached.
Looking forward & the future of AquaGen
While boxes of water & druglike compounds are an important starting point, they are only a small part of the equation of drug discovery. Through this work, we convinced ourselves that there is a compelling alternative to black box prediction, which has won over so much of ML. Gray box prediction promises to create more nuanced and useful answers to complex questions, and we believe that through this approach, much larger scale problems can potentially be tackled: lipophilicity, membrane permeability, or absolute binding free energy (ABFE). For such tasks, grounding and interpretability may be critical for the development of new drugs.
As a parting thought, while AquaGen is a state-of-the-art Boltzmann sampler, scaling experiments demonstrate that there is still a hard-to-close gap relative to MD in terms of exact structural and energetic accuracy. We have lots of ideas: model architectures can be improved, generative frameworks are still an active area of research, and we have a lot more data and labels that could be used for auxiliary prediction tasks that are currently sitting idle. Yet, ultimately we don’t believe that frameworks like AquaGen will replace MD entirely. Instead, they can be used hand-in-hand, complementing each other while delivering order-of-magnitude speed gains.
Imagine trying to screen 10,000 compounds for some property. A chemist can turn to the latest version of AquaGen to rapidly produce gray box predictions of this property, along with uncertainty estimates. Many such compounds will fall within the desired property range. Those with low AquaGen uncertainty are prime targets for bench-scale tests, while those with high uncertainties are good candidates for further MD refinements. Having full-atom samples, AquaGen predictions that challenge the chemist’s intuition, built up over years of training, can be inspected in atomic space to isolate the physics underpinning the prediction. This is how, jointly, and in the hands of experts, MD and generative models like AquaGen will accelerate drug discovery.
As a colleague likes to say, “let’s solve MD with MD!”
This post is part of “Inside Valence”, a series where you’ll get a behind-the-scenes look at our research, exploring new ways to predict, explain, and ultimately decode biology. If this resonates, consider subscribing!
 |
|
 | A guest post by
|
Realizing the importance of solvent for the dynamic of biopolymers is crucial. Finally some physics-principled thinking. Great work.